

Vers une meilleure compréhension des origines des variabilités phénotypiques du syndrome de Usher de type 3
- 10 avr.
- 3 min de lecture
Des travaux menés par les chercheurs de l’Institut reConnect, fondation sous l’égide de l’Institut Pasteur, révèlent que l’interaction compensatoire entre les protéines associées à la surdité, clarine-1 et clarine-2, gouverne la variabilité des troubles de l'audition chez les personnes concernées par le syndrome de Usher 3. L’étude, publiée le 9 avril 2026 dans Advanced Science, a été menée à l’Institut de l’Audition (Centre Institut Pasteur, Inserm, CNRS) par Maureen Wentling, doctorante, sous la co-direction de Sedigheh Delmaghani et Aziz El Amraoui, au sein de l’équipe « Déficits sensoriels progressifs, pathophysiologie et thérapie ». Les chercheurs ont montré pour la première fois le rôle essentiel et complémentaire des protéines clarine-1 et clarine-2 dans la fonction auditive, ouvrant de nouvelles perspectives thérapeutiques pour les surdités héréditaires, dont le syndrome d’Usher type 3 (USH3).
Une maladie rare aux manifestations variables
Le syndrome de Usher est une maladie rare qui constitue la première cause de surdicécité d’origine héréditaire, touchant 1 personne sur 20.000 en Europe. Il existe trois formes (types 1, 2 et 3), qui diffèrent par la sévérité de la surdité, l’atteinte de l’équilibre et l’âge d’apparition des troubles visuels. Le type 3, se caractérise par une surdité progressive et une grande variabilité entre les personnes concernées par ce syndrome, même lorsque la mutation génétique est identique. Cette variabilité restait jusqu’ici mal comprise.
Une interaction clé entre deux protéines
Dans cette étude, les chercheurs de l’Institut reConnect montrent que ce syndrome dépend en réalité d’un réseau de gènes. En particulier, le niveau de fonctionnement de la protéine clarine-2 module la sévérité des mutations du gène CLRN1, ce qui explique la variabilité phénotypique marquée chez les personnes concernées par USH3 porteurs de mutations identiques.
Ces recherches révèlent que les protéines clarine-1 et clarine-2 agissent en coopération. Ces deux protéines jouent un rôle essentiel dans les cellules ciliées de l’oreille interne, qui transforment les sons en signaux électriques. Lorsque ces deux protéines sont absentes, c'est toute la machinerie de la transduction des sons qui s'effondre. En particulier, les structures portées aux extrémités des cellules sensorielles auditives sont désorganisées, et les neurones impliqués dans l’audition subissent une dégénérescence progressive.
"Cette étude apporte une avancée majeure des mécanismes moléculaires sous-jacents à la surdité progressive, en identifiant pour la première fois le rôle compensatoire et essentiel des protéines clarine-1 et clarine-2 dans l’audition. Elle redéfinit la surdité associée à CLRN1 comme une maladie dépendante d'un réseau moléculaire, et ouvre ainsi de nouvelles orientations thérapeutiques pour les personnes concernées par le syndrome d’USH3", affirme Sedigheh Delmaghani, chercheuse à l'Institut reConnect (Centre Institut Pasteur/Inserm/CNRS).
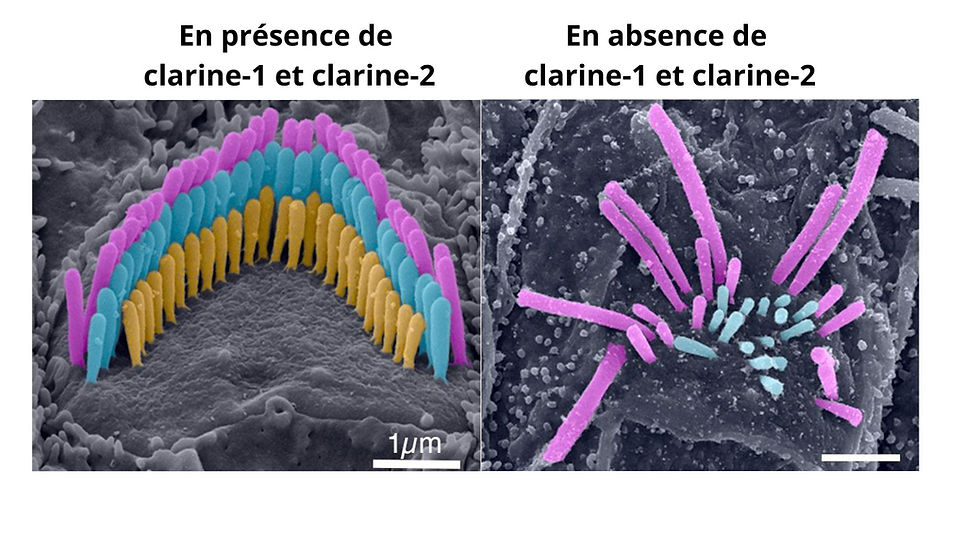
Images en microscopie électronique à balayage illustrant la désorganisation totale des structures portées aux extrémités des cellules sensorielles auditives en l’absence des gènes codant pour clarine-1 et clarine 2, comparée à une cellule normale.
Vers un meilleur dépistage
Cette recherche ouvre des perspectives pour une meilleure prise en charge de la surdité pour les personnes concernées par ce syndrome. L’objectif reste d’agir le plus tôt possible et de préserver au maximum l’audition. En montrant que la surdité de Usher de type III dépend d’un véritable réseau de gènes, cette découverte ouvre la voie à une nouvelle approche diagnostique et thérapeutique.
"Cette étude recommande donc d'inclure systématiquement le gène CLRN2 dans les analyses génétiques des patients USH3, car ses mutations pathogènes aggravent significativement la perte auditive. Par ailleurs, une implantation cochléaire réalisée avant la dégénérescence neuronale pourrait être une piste pour préserver l'audition chez les personnes qui portent des mutations dans CLRN1 et CLRN2, en stimulant directement les neurones auditifs", explique Sedigheh Delmaghani.
Référence :
M.Wentling, A.Yakhlef Sanchez, N.Thelen, et al. “Compensatory Interplay Between Clarin-1 and Clarin-2 Deafness-Associated Proteins Governs Phenotypic Variability in Hearing.” Advanced Science (2026): e21853. https://doi.org/10.1002/advs.202521853


